научное издание МГТУ им. Н.Э. Баумана
НАУКА и ОБРАЗОВАНИЕ
Издатель ФГБОУ ВПО "МГТУ им. Н.Э. Баумана". Эл № ФС 77 - 48211. ISSN 1994-0408
# 10, октябрь 2012
DOI: 10.7463/1012.0479228
УДК 621.039.587-03
Россия, Федеральное государственное автономное образовательное учреждение высшего профессионального образования
«Национальный исследовательский технологический университет «МИСиС»
julianna878@rambler.ru
kaloshkin@misis.ru
vvch@misis.ru
mvgorshenkov@gmail.com
Введение
Полимерматричные композиты являются перспективными материалами для использования в различных областях техники, в том числе и в устройствах, работающих в экстремальных условиях.
Однако, полимерные композиты пока не получили широкого применения в качестве радиационно-стойких и радиационно-защитных материалов. Значительная часть задач радиационной защиты может быть надлежащим образом решена с использованием металлических материалов [1]. Вместе с тем, существуют задачи, для эффективного решения которых необходимо существенное снижение удельной массы радиационно-защитных изделий. Очевидно, что решение этих проблем возможно только на основе полимерных материалов.
В данной работе решается задача построения комбинированной защиты на полимерной основе, обеспечивающей существенное уменьшение дозы облучения (нейтронного, γ-излучения), воздействующей на полупроводниковые приборы или персонал, находящийся в условиях высокого радиационного фона. В качестве матричного материала выбран радиационно-стойкий и биологически инертный полимер: сверхвысокомолекулярный полиэтилен (СВМПЭ). Сверхвысокомолекулярный полиэтилен – полиэтилен с молекулярной массой более 106 г/моль. Сверхвысокая молекулярная масса этого полимера определяет его уникальные физико-механические свойства, резко отличающие его от всех других марок полиэтилена. В частности, СВМПЭ обладает: а) повышенной жесткостью и исключительно высокой ударной прочностью, б) повышенным сопротивлением к абразивному воздействию (высокой износостойкостью), г) низким коэффициентом трения, сравнимым с коэффициентом трения для фторопластов, д) высокой стойкостью в агрессивных средах (коррозионной стойкостью) и повышенной стойкостью, е) возможностью эксплуатации при низких температурах (высокой морозостойкостью), ж) способностью к волокнообразованию и возможностью получения сверхпрочных нитей, превышающих по своим прочностным показателям нити из всех известных материалов. В целом, СВМПЭ можно определить как конструкционный полимерный материал с уникальными физико-механическими свойствами для разнообразных областей применения.
В качестве защищающих от γ-квантов и нейтронов наполнителей нами выбраны металлический вольфрам и карбид бора.
Для обеспечения максимальной радиационной защиты требуются большие степени наполнения полимера обеспечивающими радиационнозащитные свойства наполнителями.
Поскольку для обеспечения оптимальных радиационнозащитных характеристик композитов необходимо, прежде всего, обеспечить максимально равномерное распределение вольфрама и бора по объему материала, нами предложено использовать наполнители субмикронного и нанодисперсного размера. Для обеспечения равномерного распределения наполнителей в матрице нами предлагается отказаться от жидкофазных методов формирования полимерных композитов, негативным свойством которых является практически неизбежное формирование агломератов частиц, особенно в случае субмикронного и неразмерного наполнителя. Нами предлагается успешно опробованный ранее твердофазный метод переработки СВМПЭ.
Методика эксперимента
Для проведения структурных анализов композитов, а также для оценки степени кристалличности полимерной матрицы, важной характеристики композита, влияющей на прочность материала и его физико-механические свойства, были использованы методы рентгеноструктурного анализа и дифференциальной сканирующей калориметрии. Цель испытаний состояла в определении влияния наполнителей, термообработок, проводимых при различных технологических операциях, и механоактивационного воздействия на степень кристалличности полимерной матрицы.
1. Рентгеноструктурный анализ
Для определения фазовых превращений, протекающих в композиционных материалах на основе СВМПЭ в процессе твердофазной деформационной обработки, был проведен рентгеновский дифракционный анализ с использованием многофункционального дифрактометра RigakuUltimaIV (CoKα-излучение, длина волны 0,15178 нм). Внешний вид дифрактометра представлен на рисунке 1. Фокусировка осуществлялась по методу Брегга-Брентано с двумя щелями Соллера.

Рисунок 1 - Внешний вид дифрактометра RigakuUltimaIV
На рисунке 2а представлен рентгеновский спектр исходного СВМПЭ.
После механоактивационной обработки в течение 90 мин в высокоэнергетичном шаровом механоактиваторе АПФ-3 происходит смещение положений дифракционных максимумов кристаллической фазы в сторону больших углов (рисунок 2б).
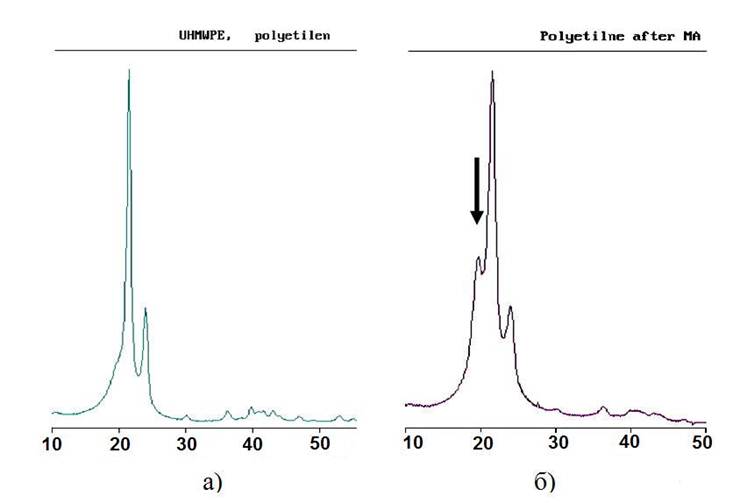
Рисунок 2 - Рентгеновский спектр чистого СВМПЭ (а) и образца СВМПЭ обработанного в механоактиваторе в течение 90 мин (б)
Рентгеновский спектр порошков СВМПЭ и карбида бора после совместной механической обработки представлен на рисунке 3.

Рисунок 3 - Рентгеновский спектр композиционного порошка СВМПЭ + 18 масс. % B4С
Для определения возможных фазовых превращений в порошках композиционных материалов при термопрессовании был проведен рентгеновский дифракционный анализ образцов, компактированных при температуре 160 °С и давлении 60 МПа.
2.Сканирующая электронная микроскопия
Изучение микроструктуры композиционных материалов методом сканирующей электронной микроскопии проводилось с помощью сканирующего электронного микроскопа JEOLJSM 6610LV (рисунок 4) при различных напряжениях в диапазоне от 5 до 20 кВ. Перед исследованием образцы полимерных композитов покрывались тонким слоем платины на установке магнетронного напыления. Исследование проводилось в режимах упруго-отраженных и вторичных электронов. Для исследования более тонких особенностей материала были получены изображения в режиме сбора вторичных электронов.

Рисунок 4 - Внешний вид сканирующего электронного микроскопа JEOLJSM 6610LV
Для проведения микроскопических исследований производился отбор проб исходных материалов и готовых композиционных порошков, а из компактированных образцов вырезался фрагмент, срез которого исследовался в микроскопе. В случае, когда срез не давал представления о структуре материала, использовался метод получения хрупких сколов путем замораживания образца полимера в жидком азоте с последующим его хрупким разрушением.
В связи с тем, что микрофотографии частиц карбида бора показали сильный разброс размеров порошинок, было решено провести измерение распределения частиц по размерам методом лазерной дифракции с использованием лазерного анализатора частиц FRITSCH Аnalysette 22.
3 Дифференциальная сканирующая калориметрия
В данной работе использовался дифференциальный сканирующий калориметр DSC 204 F1 со следующими техническими характеристиками: интервал рабочих температур от -180 до 600 ºС, скорость сканирования при нагревании или охлаждении 0.001-100 К/мин, предел чувствительности 1.0 μW; внешний вид калориметра представлен на рисунке 10.

Рисунок 10 - Внешний вид дифференциального сканирующего калориметра DSC
Исследование композиционных материалов на основе СВМПЭ проводили следующим образом: фрагмент образца композита (или навеску композиционного порошка) загружали в алюминиевый тигель, второй аналогичный тигель оставляли пустым, он служил в качестве эталонного образца, после взвешивания оба тигля устанавливали в камеру калориметра.
Для предотвращения окисления датчиков и отвода возможных продуктов горения производилась непрерывная продувка камеры аргоном. Температурная программа проведения исследований была следующей: сначала образец нагревали до 180 0С со скоростью 10 К/мин, затем охлаждали до 35 0С и повторно нагревали до 180 0С с той же скоростью. Все процессы, протекающие в ходе нагрева и охлаждения, автоматически регистрировались с помощью компьютерного программного обеспечения.
Первый нагрев производился для получения информации о том, в каком состоянии находится материал после того или иного вида обработки (механоактивация, термопрессование и.т.д.), при этом, после нагрева выше температуры плавления, происходит стирание предыстории, связанной с изменениями размеров ламеллярных кристаллов и их дефектностью, и при повтором нагреве можно определить обратимость процессов, происходящих в материале.
Результаты и обсуждение
1.Рентгеноструктурный анализ
На рисунке 2а представлен рентгеновский спектр исходного СВМПЭ. СВМПЭ является аморфно-кристаллическим полимером, кристаллическая фаза имеет орторомбическую решетку с периодами решетки a = 0,74 нм, b = 0,493 нм, c = 0,2534 нм, этой фазе соответствуют два интенсивных дифракционных максимума на рентгенограмме, с индексами 110 и 200. В результате рассеяния рентгеновских лучей аморфной фазой на дифрактограмме появляется широкое гало [2]. Центр тяжести рефлекса 110 находится при 2θ = 24.95 0, что хорошо совпадает с расчетными значениями отражений рентгеновских лучей, рассчитанных по приведенным выше параметрам решетки и уравнению Вульфа-Брегга, центр тяжести для рефлекса 200 находится также в хорошем согласии с расчетными значениями и составляет 2θ = 27,71 0.
После механоактивационной обработки в течение 90 мин в высокоэнергетичном шаровом механоактиваторе АПФ-3 происходит смещение положений дифракционных максимумов кристаллической фазы в сторону больших углов, что свидетельствует об уменьшении межплоскостного расстояния в кристаллической фазе (рисунок 2б). Положения центров тяжести рентгеновских пиков после механообработки составляют соответственно для рефлекса 110 - 2θ = 24,80 0, а для рефлекса 200 - 2θ = 27,66 0. Такой эффект, согласно [2], может быть связан с уменьшением размеров кристаллитов, что дополнительно подтверждается уширением дифракционных максимумов кристаллических областей.
Механообработка порошка СВМПЭ приводит к появлению в структуре полимера новой моноклинной фазы, присутствие которой обнаруживается по появлению одного пика при 2θ = 22,55 º, обозначенного стрелкой на рисунке 2б. Обработка рентгенограммы методом Ритвельда показывает, что в процессе механообработки происходит уменьшение количества орторомбической фазы и увеличение количества моноклинной фазы до 10 об. %, при этом количество аморфной фазы остается практически постоянным. Отсюда можно сделать вывод, что моноклинная фаза формируется в результате частичного перестроения орторомбической фазы, а также, что механическая обработка не оказывает заметного влияния на количество аморфной фазы и, соответственно, на общую степень кристалличности полимера.
Совместная механическая обработка порошков СВМПЭ и карбида бора, как видно из рисунка 3, приводит к появлению на рентгеновских дифрактограммах пиков соответствующих фазе B13С2, и линии железа, образовавшегося в результате абразивного истирания металлических шаров при механической обработке. Появления иных линий не наблюдается, что свидетельствует о протекании процесса перемешивания порошков наполнителя и матрицы без фазовых превращений.
Для определения возможных фазовых превращений в порошках композиционных материалов при термопрессовании, был проведен рентгеновский дифракционный анализ образцов, компактированных при температуре 160 °С и давлении 60 МПа. Было обнаружено, что термопрессование механообработанных порошков не приводит к изменению фазового состояния, однако моноклинная фаза, образующаяся при деформационной обработке, в процессе термопрессования полностью исчезает. Таким образом, выявлено, что моноклинная фаза в СВМПЭ является метастабильной, и компактирование при температурах, превышающих температуру плавления, приводит к ее полному исчезновению и переходу в исходную орторомбическую фазу. Роль моноклинной фазы в процессах формирования структуры СВМПЭ и композитов пока не достаточно ясна, однако следует отметить, что в ряде случаев, например, при использовании экструзии, появление данной фазы может являться желательным, так как ее образование может сопровождаться увеличением поверхностной энергии и, соответственно, улучшением технологичности материала.
2. Сканирующая электронная микроскопия
На рисунке 5 представлены микрофотографии исходных порошков карбида бора, нановольфрама в состоянии поставки. Фотографии 5А и 5Б представляют собой микросфотографии частиц нановольфрама, из которых видно, что нановольфрам представляет собой мелкодисперсный порошок с размером частиц менее 10 мкм. На рисунке 5Г представлены микрофотографии частиц карбида бора; он представляет собой мелкодисперсный порошок с размерами частиц от 0,1 до 10 мкм.


А Б


В Г
Рисунок 5 - СЭМ изображения исходного порошка нановольфрама (А) и (Б), карбид бора (В) и порошка СВМПЭ (Г)
Для определения среднего размера частиц порошка было проведено измерение распределения частиц по размерам методом лазерной дифракции с использованием лазерного анализатора частиц FRITSCHanalysette 22, результаты анализа представлены на рисунке 6. Видно, что порошок карбида бора имеет довольно широкий диапазон распределения по размерам частиц от 0,1 мкм до 10 мкм, при этом средний объемный диаметр частиц составляет 0,836 мкм.
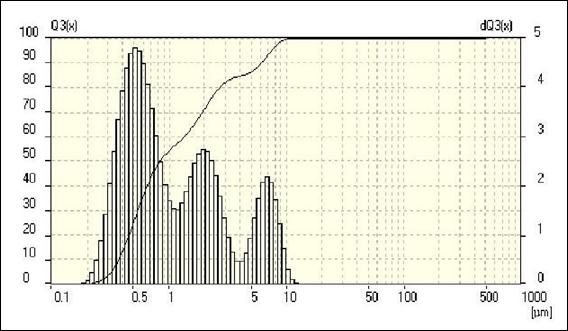
Рисунок 6 - Гранулометрический анализ исходного порошка B4С
При совместной механообработке СВМПЭ, карбида бора и нановольфрама происходит равномерное распределение карбида бора и нановольфрама по объему композиционного порошка (рисунок 7а), при этом они не проникает в объем отдельных частиц СВМПЭ, а формируют участки на поверхности частицы полимера (рисунок 7б). В процессе механического воздействия происходит дополнительное измельчение частиц карбида бора и разбрушение агломератов нановольфрама. Однако, полностью разбить агломераты вольфрама не удается, что прямым образом сказывается на механических свойствах материала, приводя к уменьшению адгезии между частицами наполнителя и полимерной матрицей.
а) |
б) |


Рисунок 7 - Микрофотографии порошка СВМПЭ с вольфрамом и карбидом бора после обработки в мельнице АПФ-3 в течение 1 часа
На рисунке 8 представлены изображения структуры среза о композиционного материала СВМПЭ + 18% W+12% B4C после компактирования. Из рисунка видно, что наполнитель распределен по матрице достаточно равномерно, однако, полностью разбить агломераты наночастиц вольфрама не удалось. Отсутствие в материале видимых дефектов структуры (поры, микротрещины и т.п.) говорит об образовании достаточно хорошего межфазного взаимодействия между полимерной матрицей и выбранными наполнителями. Однако, присутствующий на поверхности полимерных частиц наполнитель препятствует проникновению и полному спеканию полимерных частиц. При компактировании полимерные частицы образуют пространственную сетку, разделенную частицами твердых наполнителей. Прочность такой пространственной сетки может быть ниже прочности полимерной матрицы исходной при условии плохой адгезии между частицами наполнителя и матрицы. Однако подобная структура должна иметь более высокий модуль упругости и более высокий предел текучести.

Рисунок 8 - Структура композита СВМПЭ + 18% W+12% B4C после компактирования
Такая структура имеет ряд недостатков, например, более быстрое убывание механических свойств композита с увеличением степени наполнения по сравнению со структурой со случайным образом распределенными наночастицами. Теоретический предел наполнения для СВМПЭ является 33 об. %, что при используемом методе формирования композитов является практически недостижимым значением. При используемом методе смешения, предел текучести и модуль упругости композита с увеличением степени наполнения возрастают достаточно резко. Исследование композита с 80 масс. % W и 8 масс. % B4C, показывает, что вся поверхность индивидуальной частицыи полимера становится покрытой монослоем из частиц наполнителя, что приводит к катастрофическому падению механических свойств композита. Излом такого композита представлен на рисунке 9, видно, что вся поверхность полимерной частицы покрыта твердым наполнителем. Касание частиц полимера во время термопрессование происходит только в отдельных точечных участках. Сетка полимера, состоящая из таких точечных участков, является непрочной, и композит легко разрушается и имеет низкую прочность.

Рисунок 9 – Излом композита СВМПЭ+80%W+8%B4C
Таким образом, получение структуры с достаточно хорошей однородностью распределения наполнителя по матрице происходит в процессе механического воздействия, при этом наблюдается сравнительно неплохая адгезия частиц наполнителя к матрице за счет формирования сетки твердого наполнителя на поверхность полимерной частицы.
3.Дифференциальная сканирующая калориметрия
На рисунке 11 представлена кривая ДСК исходного порошка СВМПЭ (в состоянии поставки).
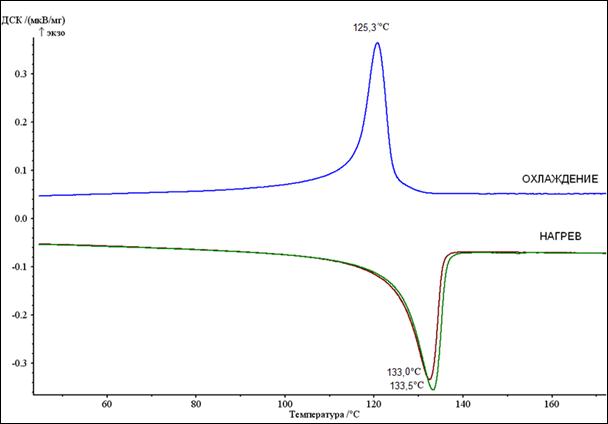
Рисунок 11 - ДСК кривая чистого СВМПЭ после термопрессования при 160 ºС
Пик, направленный вершиной вниз (эндотермический процесс) отвечает за плавление полимера, а пик, направленный вершиной вверх (экзотермический процесс), соответствует процессу кристаллизации при охлаждении. За температуру плавления (кристаллизации) в большинстве случаев принимают температуру, соответствующую вершине пика на ДСК кривой. Тепловой эффект протекающего процесса пропорционален площади, ограничиваемой линией ДСК кривой. Таким образом, определив теплоту плавления, можно провести расчет степени кристалличности исследуемого полимера по следующему соотношению:
Iст.кр.= , (1)
, (1)
где Iст.кр. – определяемая степень кристалличности, Hоб. – теплота выделившееся при плавлении, определяется площадью пика отвечающего за плавление полимера, H100 – теплота выделившееся при плавлении 100 % кристаллического полимера, для СВМПЭ H100=288 Дж/г [3]. Строго говоря, данный метод может быть применен только при условии сохранения молекулярной длины полимерных цепочек после обработки, однако, даже при изменении длин цепи, ошибка эксперимента получается небольшой и лежащей в пределах ошибки измерения.
Из рисунка 12 видно, что исходный порошок плавится при температуре 141,9 ºС, и, в соответствии с формулой (1), расчетная степень кристалличности СВМПЭ равна 72 об. %. При повторном нагреве происходит снижение температуры плавления до 133,4 ºС, а степени кристалличности до 57 об. %. Это связано с тем, что условия кристаллизации СВМПЭ при его получении отличаются от условий кристаллизации в ходе проведения измерений. Так как процесс охлаждения в калориметре идет достаточно медленно, то количество кристаллической фазы, образовавшееся при этом, значительно меньше, чем в процессе получения исходного материала.
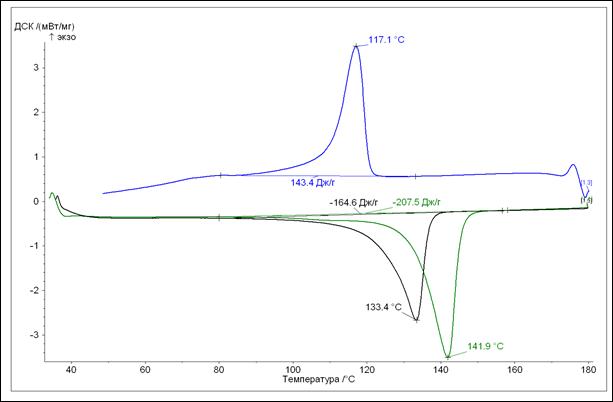 .
.
Рисунок 12 - ДСК кривая исходного порошка СВМПЭ в состоянии поставки
ДСК анализ чистого СВМПЭ после термопрессования не выявил существенных различий в поведении полимера при первом и повторном нагреве (рисунок 11). Рассчитанная по тепловому потоку при первом нагреве степень кристалличности равна 58,4 об. %, отличие между Iст.кр монолитного образца и Iст.кр при повторном нагреве исходного порошка связано с тем, что компактирование осуществляется под давлением, а давление термопрессования может сказываться на степени кристалличности СВМПЭ [4]. Однако, хотелось бы отметить некоторое снижение температуры плавления полимера, которое может быть объяснено частичным уменьшением длины цепи и, соответственно, большей подвижностью молекул полимера. При увеличении подвижности полимерных цепочек температура плавления будет снижаться, что и наблюдается в эксперименте. Наблюдаемое снижение является незначительным и не должно вести к значительным изменениям в физико-механических свойствах полимера.
Вид ДСК кривых композиционных материалов на основе СВМПЭ при использовании различных наполнителей не претерпевает заметных изменений и не отличается от приведенных на рисунках 11 и 12. В таблице 1 приведены основные результаты ДСК анализа приготовленных порошковых композиций после деформационной обработки, и монолитных образцов после термопрессования. Изменения в температуре плавления для всех образцов минимальные, в пределах погрешности измерения, отсюда следует, что введение наполнителей не приводит к заметному изменению структурного состояния композитов на основе СВМПЭ, а так же к изменению степени кристалличности. Анализ результатов теплового потока при плавлении ΔН показал его систематическое уменьшение при увеличении содержания наполнителей в полимерной матрице.
Таблица 1 - Сводная таблица основных результатов ДСК анализа
Состав | Тпик, | Тпик, | ΔН, | ΔН, | Iст.кр, % |
СВМПЭ (порошок) | 141,9 | 133,4 | 207,5 | 164,6 | 71 |
СВМПЭ+ 10% W+8%B4C (порошок) | 141,5 | 132,7 | 152,1 | 131,2 | 57 |
СВМПЭ+ 18% W+12%B4C (порошок) | 142,3 | 133,4 | 151,9 | 138,3 | 58,4 |
СВМПЭ+ 30% W+20%B4C (порошок) | 142,8 | 133,4 | 138,1 | 130,1 | 57,4 |
СВМПЭ+ 60% W+8%B4C (порошок) | 136,3 | 134,6 | 144,8 | 138,1 | 56,6 |
СВМПЭ+ 80% W+8%B4C (порошок) | 136,0 | 134,7 | 147,7 | 137,9 | 59,3 |
СВМПЭ+ 90% W+8%B4C (порошок) | 136,1 | 133,4 | 147,2 | 139,9 | 58,5 |
СВМПЭ (компакт) | 136,2 | 134,4 | 168,1 | 160,1 | 58,4 |
СВМПЭ+ 10% W+8%B4C (компакт) | 136,3 | 134,3 | 144,3 | 138,9 | 53,1 |
СВМПЭ+ 18% W+12%B4C (компакт) | 135,3 | 132,7 | 139,3 | 127,7 | 54,1 |
СВМПЭ+ 30% W+20%B4C (компакт) | 133 | 131,6 | 130,4 | 119,1 | 53,4 |
СВМПЭ+ 60% W+8%B4C (компакт) | 135,9 | 133,8 | 150,4 | 143,2 | 56,4 |
СВМПЭ+ 80% W+8%B4C (компакт) | 137 | 135 | 150,4 | 148,4 | 57,4 |
СВМПЭ+ 90% W+8%B4C (компакт) | 136,3 | 133,5 | 147,3 | 139,8 | 58,3 |
Полученные данные могут быть объяснены тем, что частицы твердого наполнителя не распределяются по матрице, а находятся только в верхнем слое полимерного материала, и не могут являться гетерогенными центрами кристаллизации [3]. Поэтому при введении наноразмерного порошка в полимерную матрицу не происходит никакого заметного увеличения степени кристалличности полимера. На рисунке 13 представлена структура полимера, выявленная при травлении СВМПЭ в смеси кислот азотной, серной и перманганата калия. Ориентированные участки – «островки» предположительно являются кристаллическими областями.
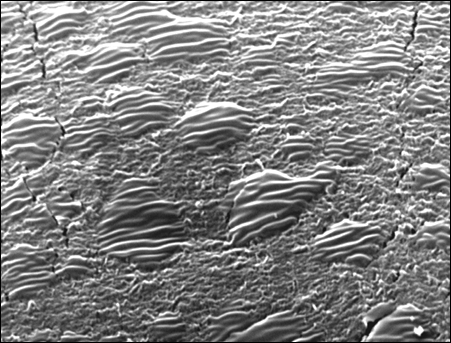
Рисунок 13 – Показана микрофотография структуры СВМПЭ после травления в смеси кислот азотной, серной и перманганата калия
Так как в исследуемом интервале температур не происходит никаких изменений в структуре используемых наполнителей, они не вносят вклад в наблюдаемые тепловые эффекты. Но в то же время, для расчета ΔН используется масса загружаемого в тигель образца, которую можно разбить на два слагаемых: масса полимерной матрицы и масса наполнителей; и чем больше содержание наполнителей, тем в образце меньше полимерной матрицы, дающей тепловой эффект. Исходя из того, что ΔН в исследуемых композитах подчиняется правилу аддитивности, для расчета степени кристалличности СВМПЭ был проведен перерасчет наблюдаемого теплового эффекта по формуле:
 , (2)
, (2)
где ΔНн – наблюдаемый тепловой эффект, Мн – степень наполнения.
Результаты расчета степени кристалличности порошковых и компактированных образцов представлены на рисунках 14 и 15. Исходный порошок СВМПЭ имеет степень кристалличности 72 об. % (рисунок 14), проведенные испытания показали, что совместная деформационная обработка в течение 90 минут порошков СВМПЭ, вольфрама и карбида бора приводит к снижению Iст.кр на 10 – 15 об. %, что объясняется частичной деструкцией СВМПЭ в процессе механообработки, в ходе которой под воздействием ударно-сдвиговых нагрузок соударяющихся шаров происходит разрыв молекулярных цепей. Еще одной причиной изменения кристалличности полимерной матрицы может быть увеличение температуры внутри барабана, что приводит к нагреву полимера и соответственно при достаточно сильном увеличении этой температуры к частичному плавлению. Частичное плавление полимерной частицы может также происходить в месте локального соударения шаров с, так как энергия шаров превращается в деформационную энергию материала и частично высвобождается в виде тепла.
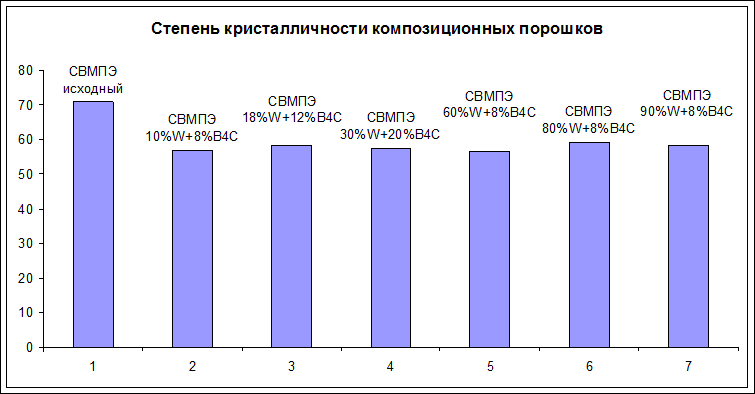
Рисунок 14 - Степень кристалличности порошковых композитов на основе СВМПЭ наполненных n-W и B4C

Рисунок 15 - Степень кристалличности композитов на основе СВМПЭ после компактирования
Существующие оценки показывают, что фоновая температура внутри барабана может достигать сотни градусов по Цельсию, а локальная температура может намного превышать фоновую. Частичной деструкцией полимерных цепей можно объяснить и небольшое изменение температуры плавления полимерной матрицы [5].
В процессе компактирования происходит нагрев полимерной матрицы выше температуры плавления, что ведет к перекристаллизация СВМПЭ, при этом, как видно из рисунка 15, степень кристалличности монолитных образов практически не отличается от степени кристалличности исходных, механообработанных порошков. Обнаруженные изменения степени кристалличности при использовании различных наполнителей незначительны, и находятся в пределах ошибки измерений. При этом для композитов можно отметить, незначительный рост степени кристалличности с возрастанием содержания наполнителей - вольфрама и карбида бора. Возможно, на поверхности частицы вольфрама все-таки играют роль гетерогенного зародышеобразователя и способствуют увеличению степени кристалличности. Однако так как все частицы сосредоточены на поверхности полимеров, то это увеличение не может являться значительным.
Заключение
На базе проведенных исследований разработана методика определения степени кристалличности композиционных материалов на основе СВМПЭ методом дифференциальной сканирующей калориметрии.
В результате проведения структурных испытаний лабораторных образцов выявлены характерные особенности получаемых механоактивационной обработкой и последующим компактированием композиционных радиационнозащитных материалов на основе СВМПЭ. Установлено, что исследуемые лабораторные образцы обладают структурой с достаточно хорошей однородностью распределения наполнителя по матрице. Лабораторные композиционные образцы характеризуются очень близкими к исходному СВМПЭ степенью кристалличности и температурой плавления, что говорит об отсутствии или незначительности процессов деструкции полимера при используемом методе получения композитов.
Работа выполнена в рамках федеральной целевой программы
«Исследования и разработки по приоритетным направлениям развития научно-технологического комплекса России на 2007-2013 годы»,
Государственный контракт от 28 апреля 2011 г. № 16.516.11.6074. Список литературы
1. Баженов С.Л., Берлин А.А., Кульков А.А., Ошмян В.Г. Полимерные композиционные материалы. Прочность и технология. Долгопрудный: Издательский дом «Интеллект», 2010. 352 с.
2. Липатов Ю.С., Шилов В.В., Гомза Ю.П., Кругляк Н.Е. Рентгенографические методы изучения полимерных систем. Киев: Науковадумка, 1982. 296 с.
3. Wunderlich B., Cormier C.M. Heat of fusion of polyethylene // Journal of Polymer Science Part A-2: Polymer Physics. 1967. Vol. 5, no. 5. P. 987-988. DOI: 10.1002/pol.1967.160050514
4. Parasnis N.C., Ramani K. Analysis of the effect of pressure on compression moulding of UHMWPE // J. of Materials Sci. Materials in medicine. 1998. Vol. 9, no. 3. P. 165-172. DOI: 10.1023/A:1008871720389
5. Jauffres D., Lame O., Vigier G., Dore F. Microstructural origin of physical and mechanical properties of ultra high molecular weight polyethylene processed by high velocity compaction // Polymer. 2007. Vol. 48, no. 21. P. 6374-6383. DOI: http://dx.doi.org/10.1016/j.polymer.2007.07.058
Публикации с ключевыми словами: карбид бора, сверхвысокомолекулярный полиэтилен, нановольфрам, структурные свойства, дисперсные частицы
Публикации со словами: карбид бора, сверхвысокомолекулярный полиэтилен, нановольфрам, структурные свойства, дисперсные частицы
Тематические рубрики:




| Авторы |
| Пресс-релизы |
| Библиотека |
| Конференции |
| Выставки |
| О проекте |
| Телефон: +7 (915) 336-07-65 (строго: среда; пятница c 11-00 до 17-00) |
|
||||
| © 2003-2024 «Наука и образование» Перепечатка материалов журнала без согласования с редакцией запрещена Тел.: +7 (915) 336-07-65 (строго: среда; пятница c 11-00 до 17-00) | |||||